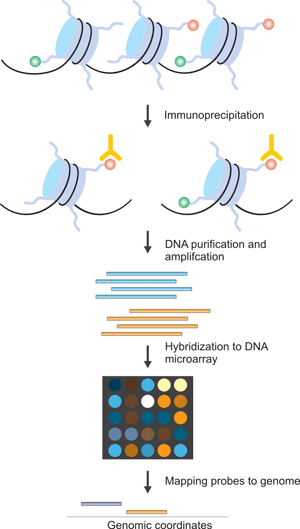
Chip Seq Histone Modification - PPT - ChIP-seq PowerPoint Presentation - ID:2695088 : For pol ii, we used a larger gap size of 1,000 to capture longer domains of pol ii binding rather than local pol ii peaks.. Chip uses antibodies to isolate a protein or modification of interest, together with any bound dna. However, little has been addressed on this issue in literature. Tag counting methods can yield high quality predictors for regression. I understand that the method to sequence tf binding sites is essentially take cells where the transcription factors are bound, freeze them, cut out the rest of the dna and sequence whats left. The six histone modifications are h3k4me1, h3k4me3, h3k27me3, h3k27ac, h3k9me3, and h3k36me3.
The gap size parameters were set to 200 for h3k4me3 and to 600 for other histone modifications as recommended. Tag counting methods can yield high quality predictors for regression. We used the macs2 peak caller (v 2.10.20130712) to identify regions of enrichment over a wide range of signal strength. Chromatin immunoprecipitation (chip) allows you to identify where histone modifications are in the genome. However, little has been addressed on this issue in literature.
Evaluation of predictability of histone marks and Pol2 ... from www.researchgate.net For pol ii, we used a larger gap size of 1,000 to capture longer domains of pol ii binding rather than local pol ii peaks. However, little has been addressed on this issue in literature. However i don't see how this method applies to. Chromatin immunoprecipitation (chip) allows you to identify where histone modifications are in the genome. The six histone modifications are h3k4me1, h3k4me3, h3k27me3, h3k27ac, h3k9me3, and h3k36me3. But now my question is related to histone modifications. Tag counting methods can yield high quality predictors for regression. We used the macs2 peak caller (v 2.10.20130712) to identify regions of enrichment over a wide range of signal strength.
Enriched regions were scored on individual.
For pol ii, we used a larger gap size of 1,000 to capture longer domains of pol ii binding rather than local pol ii peaks. It can also be utilized to identify novel biomarkers, because histone modification. The six histone modifications are h3k4me1, h3k4me3, h3k27me3, h3k27ac, h3k9me3, and h3k36me3. This can then be used to identify where the protein or modification of interest is located within the genome and its relative abundance at each location. However i don't see how this method applies to. I understand that the method to sequence tf binding sites is essentially take cells where the transcription factors are bound, freeze them, cut out the rest of the dna and sequence whats left. Some time ago i asked about what are short reads in chip seq and how come there are so many? But now my question is related to histone modifications. Enriched regions were scored on individual. This technique is widely used in stem cell research and understanding disease progression. We used the macs2 peak caller (v 2.10.20130712) to identify regions of enrichment over a wide range of signal strength. The gap size parameters were set to 200 for h3k4me3 and to 600 for other histone modifications as recommended. Chip uses antibodies to isolate a protein or modification of interest, together with any bound dna.
We used the macs2 peak caller (v 2.10.20130712) to identify regions of enrichment over a wide range of signal strength. The histone tails protruding from the nucleosome core can be modified by addition of chemical groups, mainly acetyl and methyl groups, affecting the physical accessibility of dna to the transcriptional machinery of the cell (berger, 2007; The six histone modifications are h3k4me1, h3k4me3, h3k27me3, h3k27ac, h3k9me3, and h3k36me3. Enriched regions were scored on individual. However i don't see how this method applies to.
How to Measure Histone Modification?-CUSABIO from www.cusabio.com We used the macs2 peak caller (v 2.10.20130712) to identify regions of enrichment over a wide range of signal strength. However i don't see how this method applies to. But now my question is related to histone modifications. The histone tails protruding from the nucleosome core can be modified by addition of chemical groups, mainly acetyl and methyl groups, affecting the physical accessibility of dna to the transcriptional machinery of the cell (berger, 2007; It can also be utilized to identify novel biomarkers, because histone modification. Enriched regions were scored on individual. The six histone modifications are h3k4me1, h3k4me3, h3k27me3, h3k27ac, h3k9me3, and h3k36me3. Chromatin immunoprecipitation (chip) allows you to identify where histone modifications are in the genome.
Sequence logo of identified motifs within dh sites.
We used the macs2 peak caller (v 2.10.20130712) to identify regions of enrichment over a wide range of signal strength. It can also be utilized to identify novel biomarkers, because histone modification. However i don't see how this method applies to. However, little has been addressed on this issue in literature. For pol ii, we used a larger gap size of 1,000 to capture longer domains of pol ii binding rather than local pol ii peaks. Tag counting methods can yield high quality predictors for regression. Sequence logo of identified motifs within dh sites. The gap size parameters were set to 200 for h3k4me3 and to 600 for other histone modifications as recommended. Chromatin immunoprecipitation (chip) allows you to identify where histone modifications are in the genome. Some time ago i asked about what are short reads in chip seq and how come there are so many? I understand that the method to sequence tf binding sites is essentially take cells where the transcription factors are bound, freeze them, cut out the rest of the dna and sequence whats left. Enriched regions were scored on individual. Chip uses antibodies to isolate a protein or modification of interest, together with any bound dna.
However i don't see how this method applies to. We used the macs2 peak caller (v 2.10.20130712) to identify regions of enrichment over a wide range of signal strength. Enriched regions were scored on individual. Tag counting methods can yield high quality predictors for regression. I understand that the method to sequence tf binding sites is essentially take cells where the transcription factors are bound, freeze them, cut out the rest of the dna and sequence whats left.
compbio / Epigenetic Regulation from compbio.pbworks.com This technique is widely used in stem cell research and understanding disease progression. The gap size parameters were set to 200 for h3k4me3 and to 600 for other histone modifications as recommended. Sequence logo of identified motifs within dh sites. However i don't see how this method applies to. Tag counting methods can yield high quality predictors for regression. However, little has been addressed on this issue in literature. Chromatin immunoprecipitation (chip) allows you to identify where histone modifications are in the genome. We used the macs2 peak caller (v 2.10.20130712) to identify regions of enrichment over a wide range of signal strength.
This can then be used to identify where the protein or modification of interest is located within the genome and its relative abundance at each location.
For pol ii, we used a larger gap size of 1,000 to capture longer domains of pol ii binding rather than local pol ii peaks. We used the macs2 peak caller (v 2.10.20130712) to identify regions of enrichment over a wide range of signal strength. The histone tails protruding from the nucleosome core can be modified by addition of chemical groups, mainly acetyl and methyl groups, affecting the physical accessibility of dna to the transcriptional machinery of the cell (berger, 2007; But now my question is related to histone modifications. This technique is widely used in stem cell research and understanding disease progression. Some time ago i asked about what are short reads in chip seq and how come there are so many? Enriched regions were scored on individual. Chromatin immunoprecipitation (chip) allows you to identify where histone modifications are in the genome. Chip uses antibodies to isolate a protein or modification of interest, together with any bound dna. Tag counting methods can yield high quality predictors for regression. However, little has been addressed on this issue in literature. However i don't see how this method applies to. It can also be utilized to identify novel biomarkers, because histone modification.